
Ligands
GG1_A_2001
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
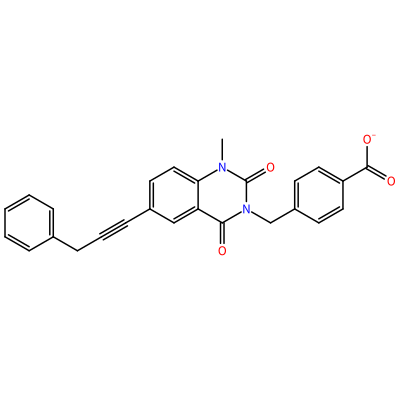
GG1_B_2002
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
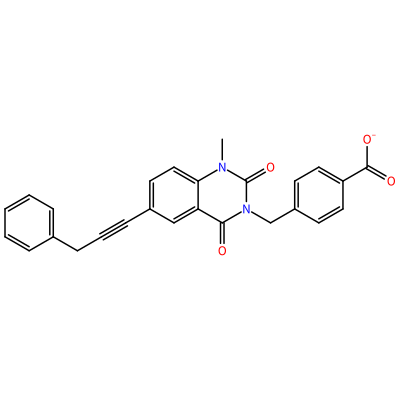
GG1_C_2003
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
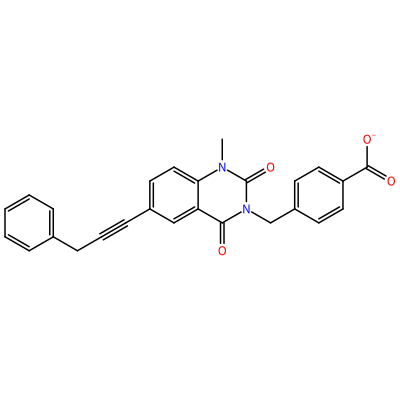
GG1_D_2004
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
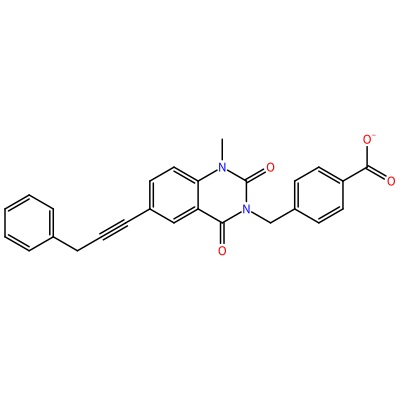
GG1_E_2005
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
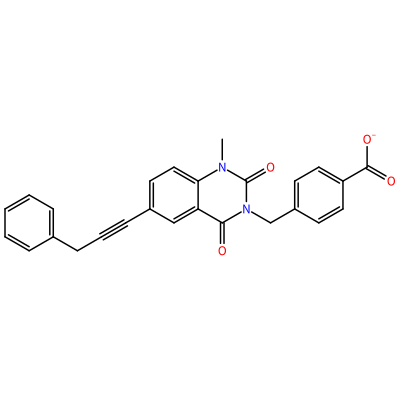
GG1_F_2006
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
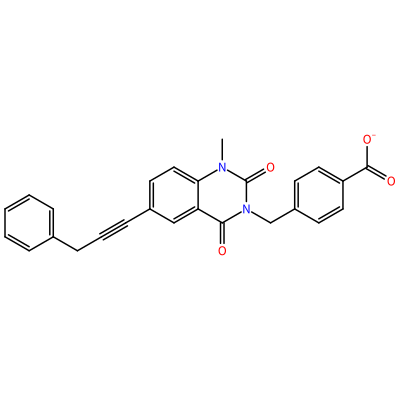
GG1_G_2007
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
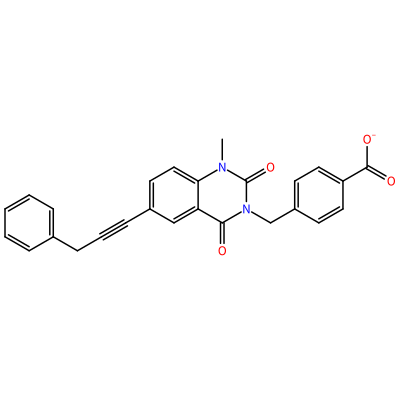
GG1_H_2008
O=C1N(C)c2ccc(C#CCc3ccccc3)cc2C(=O)N1Cc4ccc(cc4)C(=O)[O-]
HAE_C_3001
ONC(=O)C
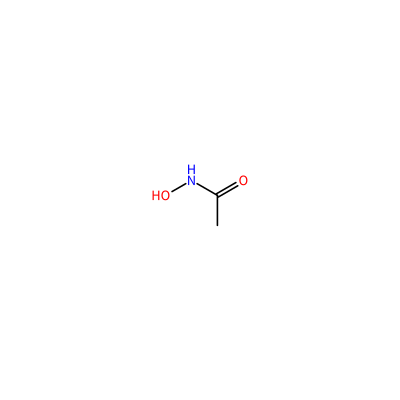
HAE_D_3002
ONC(=O)C
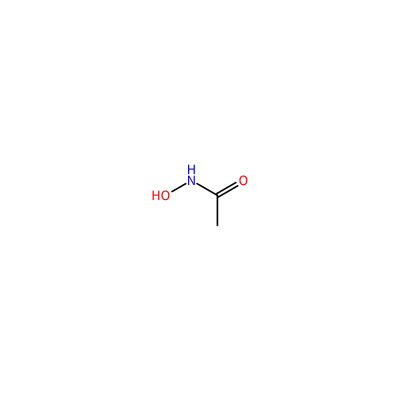
HAE_E_3003
ONC(=O)C
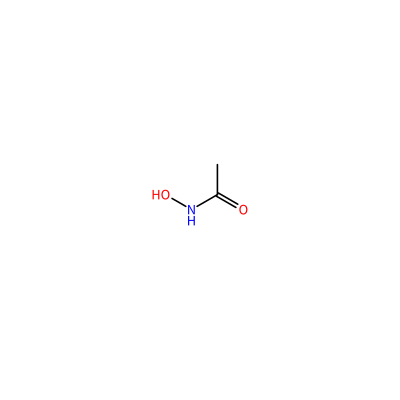
HAE_F_3004
ONC(=O)C
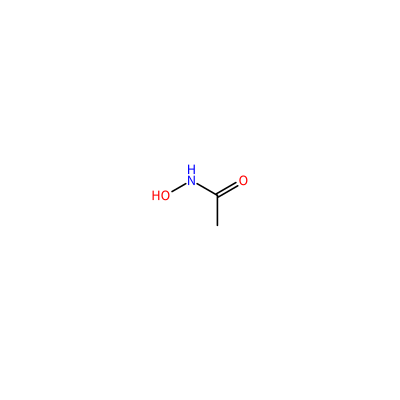
CA_A_4003
[Ca+2]

CA_A_4004
[Ca+2]

CA_A_4005
[Ca+2]

CA_B_4008
[Ca+2]

CA_B_4009
[Ca+2]

CA_B_4010
[Ca+2]

CA_C_4013
[Ca+2]

CA_C_4014
[Ca+2]

CA_C_4015
[Ca+2]

CA_D_4018
[Ca+2]

CA_D_4019
[Ca+2]

CA_D_4020
[Ca+2]

CA_E_4023
[Ca+2]

CA_E_4024
[Ca+2]

CA_E_4025
[Ca+2]

CA_F_4028
[Ca+2]

CA_F_4029
[Ca+2]

CA_F_4030
[Ca+2]

CA_G_4033
[Ca+2]

CA_G_4034
[Ca+2]

CA_G_4035
[Ca+2]

CA_H_4038
[Ca+2]

CA_H_4039
[Ca+2]

CA_H_4040
[Ca+2]

ZN_A_4001
[Zn+2]

ZN_A_4002
[Zn+2]

ZN_B_4006
[Zn+2]

ZN_B_4007
[Zn+2]

ZN_C_4011
[Zn+2]

ZN_C_4012
[Zn+2]

ZN_D_4016
[Zn+2]

ZN_D_4017
[Zn+2]

ZN_E_4021
[Zn+2]

ZN_E_4022
[Zn+2]

ZN_F_4026
[Zn+2]

ZN_F_4027
[Zn+2]

ZN_G_4031
[Zn+2]

ZN_G_4032
[Zn+2]

ZN_H_4036
[Zn+2]

ZN_H_4037
[Zn+2]

Pockets
calculating Pockets, this may take a minute
Protoss is a fully automated hydrogen prediction tool for protein-ligand complexes.
It adds missing hydrogen atoms to protein structures (PDB-format) and detects reasonable protonation states,
tautomers, and hydrogen coordinates of both protein and ligand molecules. Protoss investigates hydrogen bonds,
metal interactions and repulsive atom contacts for all possible states and calculates an optimal hydrogen bonding
network within these degrees of freedom. Furthermore, alternative conformations or overlapping entries that might
be annotated in the original protein structure are removed, as they could disturb the analysis of molecular
interactions.1,2
1. Lippert, T.; Rarey, M., Fast automated placement of
polar hydrogen atoms in protein-ligand complexes. J Cheminform 2009, 1 (1), 13. DOI:
https://doi.org/10.1186/1758-2946-1-13
2. Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M.,
Protoss: a holistic approach to predict tautomers and protonation states
in protein-ligand complexes. J Cheminform 2014, 6, 12. DOI:
https://doi.org/10.1186/1758-2946-6-12
DoGSiteScorer is a grid-based method that uses a Difference of Gaussian filter to detect potential binding pockets1 - solely based on the 3D structure of the protein - and splits them into subpockets.
Global properties, describing the size, shape and chemical features of the predicted (sub)pockets are calculated. Per default, a simple druggability score is provided for each (sub)pocket, based on a linear combination of the three descriptors describing volume, hydrophobicity and enclosure. Furthermore, a subset of meaningful descriptors is incorporated in a support vector machine (libsvm) to predict the (sub)pocket druggability score (values are between zero and one). The higher the score the more druggable the pocket is estimated to be.2
1. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M.,
Analyzing the topology of active sites: on the prediction of pockets and subpockets.
J Chem Inf Model 2010, 50 (11), 2041-52. DOI:
https://doi.org/10.1021/ci100241y
2. Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M.,
Combining global and local measures for structure-based druggability predictions.
J Chem Inf Model 2012, 52 (2), 360-72. DOI:
https://doi.org/10.1021/ci200454v
DoGSite31 is a grid-based method which uses a Difference of Gaussian
filter to detect potential binding pockets - solely based on the 3D structure of the macromolecules
(protein and nucleic acid chains with > 50 atoms are considered) - and splits
them into subpockets. It is a reimplementation of the DoGSite algorithm2,
3
with optimized parameters and an optional method to make use of a ligand bias.
Global properties, describing the size, shape and chemical features of the predicted (sub)pockets are calculated.
1. Graef, J.; Ehrt, C.; Rarey, M.,
Binding Site Detection Remastered: Enabling Fast, Robust, and Reliable Binding Site Detection
and Descriptor Calculation with DoGSite3.
J Chem Inf Model 2023, 63 (10), 3128–3137. DOI:
https://doi.org/10.1021/acs.jcim.3c00336
2. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M.,
Analyzing the topology of active sites: on the prediction of pockets and subpockets.
J Chem Inf Model 2010, 50 (11), 2041-52. DOI:
https://doi.org/10.1021/ci100241y
3. A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann, M. Rarey.
Combining global and local measures for structure-based druggability predictions.
J. Chem. Inf. Model. 2012,52,360-372.
PoseView automatically creates two-dimensional diagrams of complexes with known
3D structure according to chemical drawing conventions.
1,2,3
Directed bonds between protein and ligand are drawn as dashed lines
and the interacting protein residues and the ligand are visualized as structure
diagrams. Hydrophobic contacts are represented more indirectly through
spline sections highlighting the hydrophobic parts of the ligand and the label
of the contacting residue. The generation of structure diagrams and their
layout modifications are based on the library 2Ddraw. 4 Interactions between
the molecules are estimated by a builtin interaction model that is based on atom
types and simple geometric criteria.
1. Stierand, K.; Maass, P. C.; Rarey, M.,
Drawing the PDB - Protein-Ligand Complexes in two Dimensions.
Medicinal Chemistry Letters 2010, 1 (9) 540-545. DOI:
https://doi.org/10.1021/ml100164p
2. Stierand, K.; Rarey, M.,
From Modeling to Medicinal Chemistry: Automatic Generation of Two-Dimensional
Complex Diagrams. ChemMedChem 2007, 2 (6), 853-860. DOI:
https://doi.org/10.1002/cmdc.200700010
3. Stierand, K.; Maass, P. C.; Rarey, M.,
Molecular complexes at a glance: automated generation of two-dimensional
complex diagrams. Bioinformatics 2006, 22 (14), 1710-6. DOI:
https://doi.org/10.1093/bioinformatics/btl150
4. Fricker, P. C.; Gastreich, M.; Rarey, M.,
Automated drawing of structural molecular formulas under constraints.
J Chem Inf Comput Sci 2004, 44 (3), 1065-78. DOI:
https://doi.org/10.1021/ci049958u
PoseEdit 1 uses the PoseView algorithm
and the InteractionDrawer JavaScript library (https://github.com/rareylab/InteractionDrawer) for the fully automatic drawing of highly interactive 2D ligand interaction diagrams.
Structures are depicted according to IUPAC recommendations.
Calculated interactions (hydrogen bonds, cation-pi interactions, pi-stackings, ionic interactions, and metal interactions)
of the ligand with nucleic acids, amino acids, and metals are depicted as colored dashed lines.
Hydrophobic contacts are visualized by labeled green splines.
The binding sites and the interactions occurring within were calculated by Protoss, DoGSite3, and GeoMine.
1. Diedrich, K.; Krause, B.; Berg, O.; Rarey, M., PoseEdit: enhanced ligand binding mode communication by interactive 2D diagrams. J Comput Aided Mol Des 2023. DOI:
https://doi.org/10.1007/s10822-023-00522-4
SIENA has been developed for the automated assembly and preprocessing of protein binding site ensembles.
Starting with a single binding site, SIENA searches the PDB for alternative conformations of the same or sequentially closely related binding sites.
The method is based on an indexed database for identifying perfect k-mer matches and
a new algorithm for the detection of protein binding site conformations.
Furthermore SIENA provides a variety of different filters for pruning the resulting conformational ensemble
to meet a user’s case specific requirements. This involves
a new algorithm for the interaction-based selection of binding site conformations as well as an RMSD-based
clustering for ensemble reduction.
SIENA provides the user with a sequence alignment of the binding site as well
as superimposed PDB structures which are, apart from the transferred coordinates, equal to the
original files from the PDB and thus contain all structural details and further information.
1,2
1. Bietz, S.; Rarey, M., ASCONA: Rapid Detection and Alignment of
Protein Binding Site Conformations. J Chem Inf Model 2015, 55 (8), 1747-56. DOI:
https://doi.org/10.1021/acs.jcim.5b00210
2. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of Selective Protein Binding
Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI:
https://doi.org/10.1021/acs.jcim.5b00588
HyPPI classifies a protein-protein complex concerning its interaction type into permanent, transient or crystal artifact. Permanent protein-protein complexes are only stable in their complexed state and the subunits would denature upon complex dissociation. Transient protein-protein complexes are stable in the complexed as well as in the monomeric form depending on the necessary function of the complex. Crystal artifacts have no biological function and are artifically formed during the crystallization process.
The discrimination is performed using two characteristics of the protein-protein complex, the hydrophobicity of the interface (ΔGHydrophobic) and the quotient of interface area ratios (IF-quotient). ΔGHydrophobic represents the energy emerging exclusively from the hydrophobic effect upon binding of two protein subunits and was calculated according to the desolvation term of the HYDE scoring function1. The IF-quotient takes the symmetry of the protein-protein interface into account.
1
1. Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey,
M., A consistent description of HYdrogen bond and Dehydration energies
in protein-ligand complexes: methods behind the HYDE scoring function.
J Comput Aided Mol Des 2013, 27 (1), 15-29. DOI:
https://doi.org/10.1007/s10822-012-9626-2
EDIA expects the EDM to cover every atom of interest in the pdb file. Therefore, the electron density maps are calculated to cover the complete pdb file by means of the GEMMI tool sf2map2.
1. Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M., Estimating Electron Density Support for Individual Atoms and Molecular Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI: https://doi.org/10.1021/acs.jcim.7b00391
2. Wojdyr, M., GEMMI: A library for structural biology. Journal of Open Source Software (2022), 7(73), 4200. DOI: https://doi.org/10.21105/joss.04200 EDIA
METALizer predicts the coordination geometry of metals in metalloproteins.
Potential coordination geometries of metals are matched onto the found
metal interactions in the examined structure.
The predicted coordination geometries and the observed metal
interaction distances can be compared interactively to statistics calculated based on
the PDB.
Furthermore, METALizer is combined with other tools in the ProteinsPlus server:
Using SIENA1, ensembles of proteins
with sequentially and structurally closely related metal binding
sites can be
retrieved from the PDB, superimposed and visualized.
This allows the comparison of the predicted coordination geometries
and metal interaction distances
to statistics calculated only on related metal binding sites.
Furthermore, different binding modes of ligands to the metal and of the metal
within the protein can be explored.
Another option is the EDIA2 filter to detect
atoms that are poorly supported by electron density. These are then excluded
from the METALizer analysis.
1. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of
Selective Protein Binding Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI:
https://doi.org/10.1021/acs.jcim.5b00588
2. Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M.,
Estimating Electron Density Support for Individual Atoms and Molecular
Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI:
https://doi.org/10.1021/acs.jcim.7b00391
ActivityFinder1 links bioactivity data, in this case from ChEMBL, and structural data from the Protein Data Bank automatically at the atomic level.
ActivityFinder data can now be inspected directly in ProteinsPlus from the precomputed ChEMBL tables. You can also query it using the REST API at https://proteins.plus/api/v2/activityfinder/.
For a detailed explanation of how to query the REST API, please consult the GitHub.
For the database used to query, consult the Uni Hamburg FDR, and for data of the complete PDB, consult the respective entry at the FDR.
1. Ehmki, E. S. R.; Gutermuth, T.; Harren, T.; Kurtz, S.; Rarey, M.,
ActivityFinder: Toward the Fully Automatic Integration of Structural and Binding Affinity Data.
Journal of Chemical Information and Modeling 2026 66 (2), 1013-1034. DOI:
https://doi.org/10.1021/acs.jcim.5c02505
LifeSoaks calculates solvent channels and their corresponding
bottleneck radii by construcing a three dimensional Voronoi
diagram.
You can use it to compute a bottleneck radius for a complete
crystal structure as well as a ccp4 density file storing
local bottleneck radii in order to analyze the structure with
respect to specific regions of interest.
When changing the cut-off radius, the displayed
grid changes as well. For the current cut-off, the visualized
grid represents the space in which a sphere with that radius
could roll around freely on an infinite path through the crystal.
When the protein strongly reaches outside its unit cell, it might
be necessary to visualize several asymmetric unit copies, which is
currently not supported in this viewer. We recommend downloading
the ccp4 file in these cases and loading it with more
sophisticated visualization programs.
1. Pletzer-Zelgert, J.; Ehrt, C.; Fender, I.; Griewel, A.; Flachsenberg, F.; Klebe, G.; Rarey, M.,
LifeSoaks: a tool for analyzing solvent channels in protein crystals and obstacles for soaking experiments.
Acta Cryst. 2023, D79, 837-856. DOI:
https://doi.org/10.1107/S205979832300582X
WarPP places water molecules for the active site of a given PDB file.1
Based on interaction geometries previously derived from protein crystal structures potential water
positions are generated for free interaction directions (lone pairs of acceptors or hydrogen atoms of donors).
Every small molecule present in the PDB structure is used to generate an active site in a 6.5 Å radius of each atom.
Within these active sites water molecules are placed.
In the corresponding publication we showed that WarPP places 80% of water molecules within 1.0 Å
distance to a crystallographically observed one.
1. Nittinger, E.; Flachsenberg, F.; Bietz, S.; Lange, G.;
Klein, R.; Rarey, M., Placement of Water Molecules in Protein Structures:
From Large-Scale Evaluations to Single-Case Examples. J Chem Inf Model 2018, 58 (8),
1625-1637. DOI:
https://doi.org/10.1021/acs.jcim.8b00271
StructureProfiler was developed as an all-in-one tool to screen structures
based on selection criteria typically used upon dataset assembly for
structure-based design methods.1 Test configurations based on either the
Astex2, the Iridium HT3,
the Platinum4, or the combination of all three
criteria catalogs. (Note that real-space correlation coefficient (RSCC) as an electron density validation
criteria was replaced by EDIAm5 scoring).
1. Meyder, A.; Kampen, S.; Sieg, J.; Fahrrolfes,
R.; Friedrich, N. O.; Flachsenberg, F.; Rarey, M.,
StructureProfiler: an all-in-one tool for 3D protein structure profiling.
Bioinformatics 2019, 35 (5), 874-876. DOI:
https://doi.org/10.1093/bioinformatics/bty692
2. Hartshorn, M. J.; Verdonk, M. L.; Chessari,
G.; Brewerton, S. C.; Mooij, W. T.; Mortenson, P. N.; Murray, C. W.,
Diverse, high-quality test set for the validation of protein-ligand docking
performance. J Med Chem 2007, 50 (4), 726-41. DOI:
https://doi.org/10.1021/jm061277y
3. Warren, G. L.; Do, T. D.; Kelley, B. P.;
Nicholls, A.; Warren, S. D., Essential considerations for using protein-ligand
structures in drug discovery. Drug Discov Today 2012, 17 (23-24), 1270-81. DOI:
https://doi.org/10.1016/j.drudis.2012.06.011
4. Friedrich, N. O.; Meyder, A.; de Bruyn Kops,
C.; Sommer, K.; Flachsenberg, F.; Rarey, M.; Kirchmair, J.,
High-Quality Dataset of Protein-Bound Ligand Conformations and Its
Application to Benchmarking Conformer Ensemble Generators.
J Chem Inf Model 2017, 57 (3), 529-539. DOI:
https://doi.org/10.1021/acs.jcim.6b00613
5. Meyder, A.; Nittinger, E.; Lange, G.; Klein,
R.; Rarey, M., Estimating Electron Density Support for Individual Atoms and Molecular
Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI:
https://doi.org/10.1021/acs.jcim.7b00391
GeoMine enables textual, numerical and 3D searching with full chemical
awareness in binding sites of the entire PDB dataset.
1,2
We are aware of a problem regarding picking from the structure:
This problem does seem to only occur in Chromium-based browsers (e.g., Chrome/Edge).
Safari and Firefox are not affected.
We are working on a fix - thanks for your patience.
The tool is based on the desktop application Pelikan.3
It enables various user-defined queries for on-the-fly binding site comparison for
predicted and ligand-based binding sites in the PDB.
Data from PDB files is preprocessed and stored in a PostgreSQL database.
1. Graef, J.; Ehrt, C.; Diedrich, K.; Poppinga, M.; Ritter, N.;
Rarey, M., Searching Geometric Patterns in Protein Binding Sites and
Their Application to Data Mining in Protein Kinase Structures.
J Med Chem 2022, 65 (2), 1384-1395. DOI:
https://doi.org/10.1021/acs.jmedchem.1c01046
2. Diedrich, K.; Graef, J.; Schoning-Stierand, K.; Rarey,
M., GeoMine: interactive pattern mining of protein-ligand interfaces
in the Protein Data Bank. Bioinformatics 2021, 37 (3), 424-425. DOI:
https://doi.org/10.1093/bioinformatics/btaa693
3. Inhester, T.; Bietz, S.; Hilbig, M.; Schmidt, R.;
Rarey, M., Index-Based Searching of Interaction Patterns in Large Collections of
Protein-Ligand Interfaces. J Chem Inf Model 2017, 57 (2), 148-158. DOI:
https://doi.org/10.1021/acs.jcim.6b00561
JAMDA is a novel and fully automated protein-ligand docking tool.1
It combines the TrixX docking algorithm
2,3
with the JAMDA scoring function.4
The docking and scoring performance of JAMDA is in line with the state of the
art in the field. JAMDA has an unprecedented level of automation enabling
molecular docking for everyone on the web. Furthermore, the JAMDA scoring
function is combined with a new gradient-based optimizer achieving high
numerical stability.
Starting from a protein structure and the molecules to be docked, the structures are preprocessed and docked
automatically. The docking site can be defined by a reference ligand or by a pocket from the pockets tab (that has been either built by hand or
predicted by DoGSiteScorer5). The molecules for docking can be uploaded without any preprocessing i.e., neither
coordinates nor protonation states are required.
1. Flachsenberg, F.; Ehrt, C.; Gutermuth, T.; Rarey, M., Redocking the PDB. J Chem Inf Model 2023. DOI:
https://doi.org/10.1021/acs.jcim.3c01573
2. Schellhammer, I.; Rarey, M., TrixX: structure-based
molecule indexing for large-scale virtual screening in sublinear time.
J Comput Aided Mol Des 2007, 21 (5), 223-38. DOI:
https://doi.org/10.1007/s10822-007-9103-5
3. Henzler, A. M.; Urbaczek, S.; Hilbig, M.; Rarey, M.,
An integrated approach to knowledge-driven structure-based virtual screening.
J Comput Aided Mol Des 2014, 28 (9), 927-39. DOI:
https://doi.org/10.1007/s10822-014-9769-4
4. Flachsenberg, F.; Meyder, A.; Sommer, K.; Penner, P.;
Rarey, M., A Consistent Scheme for Gradient-Based Optimization of
Protein-Ligand Poses. J Chem Inf Model 2020, 60 (12), 6502-6522. DOI:
https://doi.org/10.1021/acs.jcim.0c01095
5. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M.,
Analyzing the topology of active sites: on the prediction of pockets and subpockets.
J Chem Inf Model 2010, 50 (11), 2041-52. DOI:
https://doi.org/10.1021/ci100241y
MicroMiner1 searches mutations in protein structure databases like the PDB. Retrieved mutant structures can be
easily analysed and compared to the wild-type through automatically superposed structures. Local mutation
sites can be filtered by RMSD to identify structurally deviations upon mutation.
The method uses the local 3D micro-environment of single residues in a query protein structure to
search for similar local environments in a protein structure database where the central query residue is mutated.
MicroMiner returns structures with similar environments that are identical in sequence -except for the
position of the mutation- and have a similar structural arrangement. These mutant structures are
automatically superposed to the query wild-type protein for easy comparison and analysis of the
structural changes of mutations. The method originates from the
ASCONA2/SIENA3 technology.
1. Sieg, J.; Rarey, M., Searching similar local 3D micro-environments in protein structure databases with
MicroMiner. Brief. Bioinform. 2023, 24 (6), bbad357. DOI:
https://doi.org/10.1093/bib/bbad357
2. Bietz, S.; Rarey, M., ASCONA: Rapid Detection and Alignment of Protein Binding Site
Conformations. J Chem Inf Model 2015, 55 (8), 1747-56. DOI:
https://doi.org/10.1021/acs.jcim.5b00210
3. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of Selective Protein
Binding Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI:
https://doi.org/10.1021/acs.jcim.5b00588
Protoss
Protoss is a fully automated hydrogen prediction tool for protein-ligand complexes.
It adds missing hydrogen atoms to protein structures (PDB-format) and detects reasonable protonation states,
tautomers, and hydrogen coordinates of both protein and ligand molecules. Protoss investigates hydrogen bonds,
metal interactions and repulsive atom contacts for all possible states and calculates an optimal hydrogen bonding
network within these degrees of freedom. Furthermore, alternative conformations or overlapping entries that might
be annotated in the original protein structure are removed, as they could disturb the analysis of molecular
interactions.1,2
1. Lippert, T.; Rarey, M., Fast automated placement of
polar hydrogen atoms in protein-ligand complexes. J Cheminform 2009, 1 (1), 13. DOI:
https://doi.org/10.1186/1758-2946-1-13
2. Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M.,
Protoss: a holistic approach to predict tautomers and protonation states
in protein-ligand complexes. J Cheminform 2014, 6, 12. DOI:
https://doi.org/10.1186/1758-2946-6-12
DoGSiteScorer
DoGSiteScorer is a grid-based method that uses a Difference of Gaussian filter to detect potential binding pockets1 - solely based on the 3D structure of the protein - and splits them into subpockets.
Global properties, describing the size, shape and chemical features of the predicted (sub)pockets are calculated. Per default, a simple druggability score is provided for each (sub)pocket, based on a linear combination of the three descriptors describing volume, hydrophobicity and enclosure. Furthermore, a subset of meaningful descriptors is incorporated in a support vector machine (libsvm) to predict the (sub)pocket druggability score (values are between zero and one). The higher the score the more druggable the pocket is estimated to be.2
1. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M.,
Analyzing the topology of active sites: on the prediction of pockets and subpockets.
J Chem Inf Model 2010, 50 (11), 2041-52. DOI:
https://doi.org/10.1021/ci100241y
2. Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M.,
Combining global and local measures for structure-based druggability predictions.
J Chem Inf Model 2012, 52 (2), 360-72. DOI:
https://doi.org/10.1021/ci200454v
Settings
DoGSite3
DoGSite31 is a grid-based method which uses a Difference of Gaussian
filter to detect potential binding pockets - solely based on the 3D structure of the macromolecules
(protein and nucleic acid chains with > 50 atoms are considered) - and splits
them into subpockets. It is a reimplementation of the DoGSite algorithm2,
3
with optimized parameters and an optional method to make use of a ligand bias.
Global properties, describing the size, shape and chemical features of the predicted (sub)pockets are calculated.
1. Graef, J.; Ehrt, C.; Rarey, M.,
Binding Site Detection Remastered: Enabling Fast, Robust, and Reliable Binding Site Detection
and Descriptor Calculation with DoGSite3.
J Chem Inf Model 2023, 63 (10), 3128–3137. DOI:
https://doi.org/10.1021/acs.jcim.3c00336
2. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M.,
Analyzing the topology of active sites: on the prediction of pockets and subpockets.
J Chem Inf Model 2010, 50 (11), 2041-52. DOI:
https://doi.org/10.1021/ci100241y
3. A. Volkamer, D. Kuhn, T. Grombacher, F. Rippmann, M. Rarey.
Combining global and local measures for structure-based druggability predictions.
J. Chem. Inf. Model. 2012,52,360-372.
Settings
SIENA
SIENA has been developed for the automated assembly and preprocessing of protein binding site ensembles.
Starting with a single binding site, SIENA searches the PDB for alternative conformations of the same or sequentially closely related binding sites.
The method is based on an indexed database for identifying perfect k-mer matches and
a new algorithm for the detection of protein binding site conformations.
Furthermore SIENA provides a variety of different filters for pruning the resulting conformational ensemble
to meet a user’s case specific requirements. This involves
a new algorithm for the interaction-based selection of binding site conformations as well as an RMSD-based
clustering for ensemble reduction.
SIENA provides the user with a sequence alignment of the binding site as well
as superimposed PDB structures which are, apart from the transferred coordinates, equal to the
original files from the PDB and thus contain all structural details and further information.
1,2
1. Bietz, S.; Rarey, M., ASCONA: Rapid Detection and Alignment of
Protein Binding Site Conformations. J Chem Inf Model 2015, 55 (8), 1747-56. DOI:
https://doi.org/10.1021/acs.jcim.5b00210
2. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of Selective Protein Binding
Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI:
https://doi.org/10.1021/acs.jcim.5b00588
Settings
PoseView
PoseView automatically creates two-dimensional diagrams of complexes with known
3D structure according to chemical drawing conventions.
1,2,3
Directed bonds between protein and ligand are drawn as dashed lines
and the interacting protein residues and the ligand are visualized as structure
diagrams. Hydrophobic contacts are represented more indirectly through
spline sections highlighting the hydrophobic parts of the ligand and the label
of the contacting residue. The generation of structure diagrams and their
layout modifications are based on the library 2Ddraw. 4 Interactions between
the molecules are estimated by a builtin interaction model that is based on atom
types and simple geometric criteria.
1. Stierand, K.; Maass, P. C.; Rarey, M.,
Drawing the PDB - Protein-Ligand Complexes in two Dimensions.
Medicinal Chemistry Letters 2010, 1 (9) 540-545. DOI:
https://doi.org/10.1021/ml100164p
2. Stierand, K.; Rarey, M.,
From Modeling to Medicinal Chemistry: Automatic Generation of Two-Dimensional
Complex Diagrams. ChemMedChem 2007, 2 (6), 853-860. DOI:
https://doi.org/10.1002/cmdc.200700010
3. Stierand, K.; Maass, P. C.; Rarey, M.,
Molecular complexes at a glance: automated generation of two-dimensional
complex diagrams. Bioinformatics 2006, 22 (14), 1710-6. DOI:
https://doi.org/10.1093/bioinformatics/btl150
4. Fricker, P. C.; Gastreich, M.; Rarey, M.,
Automated drawing of structural molecular formulas under constraints.
J Chem Inf Comput Sci 2004, 44 (3), 1065-78. DOI:
https://doi.org/10.1021/ci049958u
Settings
PoseEdit
PoseEdit 1 uses the PoseView algorithm
and the InteractionDrawer JavaScript library (https://github.com/rareylab/InteractionDrawer) for the fully automatic drawing of highly interactive 2D ligand interaction diagrams.
Structures are depicted according to IUPAC recommendations.
Calculated interactions (hydrogen bonds, cation-pi interactions, pi-stackings, ionic interactions, and metal interactions)
of the ligand with nucleic acids, amino acids, and metals are depicted as colored dashed lines.
Hydrophobic contacts are visualized by labeled green splines.
The binding sites and the interactions occurring within were calculated by Protoss, DoGSite3, and GeoMine.
1. Diedrich, K.; Krause, B.; Berg, O.; Rarey, M., PoseEdit: enhanced ligand binding mode communication by interactive 2D diagrams. J Comput Aided Mol Des 2023. DOI:
https://doi.org/10.1007/s10822-023-00522-4
Settings
Protein-protein interactions
HyPPI classifies a protein-protein complex concerning its interaction type into permanent, transient or crystal artifact. Permanent protein-protein complexes are only stable in their complexed state and the subunits would denature upon complex dissociation. Transient protein-protein complexes are stable in the complexed as well as in the monomeric form depending on the necessary function of the complex. Crystal artifacts have no biological function and are artifically formed during the crystallization process.
The discrimination is performed using two characteristics of the protein-protein complex, the hydrophobicity of the interface (ΔGHydrophobic) and the quotient of interface area ratios (IF-quotient). ΔGHydrophobic represents the energy emerging exclusively from the hydrophobic effect upon binding of two protein subunits and was calculated according to the desolvation term of the HYDE scoring function. The IF-quotient takes the symmetry of the protein-protein interface into account.1
1. Schneider, N.; Lange, G.; Hindle, S.; Klein, R.; Rarey,
M., A consistent description of HYdrogen bond and Dehydration energies
in protein-ligand complexes: methods behind the HYDE scoring function.
J Comput Aided Mol Des 2013, 27 (1), 15-29. DOI:
https://doi.org/10.1007/s10822-012-9626-2
Settings
EDIA
EDIA - the electron density score for individual atoms quantifies the electron density fit of an atom. Multiple EDIA values can be combined with the help of the power mean to compute EDIAm, the electron density score for multiple atoms to score a set of atoms such as a ligand or residue. Substructures such as residues and ligands can be automatically analyzed with EDIAm.
EDIA expects the EDM to cover every atom of interest in the pdb file. Therefore, the electron density maps are calculated to cover the complete pdb file by means of the GEMMI library tool sf2map 3.
1. Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M., Estimating Electron Density Support for Individual Atoms and Molecular Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI: https://doi.org/10.1021/acs.jcim.7b00391
2. Wojdyr, M., GEMMI: A library for structural biology. Journal of Open Source Software (2022), 7(73), 4200. DOI: https://doi.org/10.21105/joss.04200
METALizer
METALizer predicts the coordination geometry of metals in metalloproteins.
Potential coordination geometries of metals are matched onto the found
metal interactions in the examined structure.
The predicted coordination geometries and the observed metal
interaction distances can be compared interactively to statistics calculated based on
the PDB.
Furthermore, METALizer is combined with other tools in the ProteinsPlus server:
Using SIENA1, ensembles of proteins
with sequentially and structurally closely related metal binding
sites can be
retrieved from the PDB, superimposed and visualized.
This allows the comparison of the predicted coordination geometries
and metal interaction distances
to statistics calculated only on related metal binding sites.
Furthermore, different binding modes of ligands to the metal and of the metal
within the protein can be explored.
Another option is the EDIA2 filter to detect
atoms that are poorly supported by electron density. These are then excluded
from the METALizer analysis.
1. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of
Selective Protein Binding Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI:
https://doi.org/10.1021/acs.jcim.5b00588
2. Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M.,
Estimating Electron Density Support for Individual Atoms and Molecular
Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI:
https://doi.org/10.1021/acs.jcim.7b00391
Settings
ActivityFinder
ActivityFinder1 links bioactivity data, in this case from ChEMBL, and structural data from the Protein Data Bank automatically at the atomic level.
ActivityFinder data can now be inspected directly in ProteinsPlus from the precomputed ChEMBL tables. You can also query it using the REST API at https://proteins.plus/api/v2/activityfinder/.
For a detailed explanation of how to query the REST API, please consult the GitHub.
For the database used to query, consult the Uni Hamburg FDR, and for data of the complete PDB, consult the respective entry at the FDR.
1. Ehmki, E. S. R.; Gutermuth, T.; Harren, T.; Kurtz, S.; Rarey, M.,
ActivityFinder: Toward the Fully Automatic Integration of Structural and Binding Affinity Data.
Journal of Chemical Information and Modeling 2026 66 (2), 1013-1034. DOI:
https://doi.org/10.1021/acs.jcim.5c02505
MicroMiner
MicroMiner1 searches mutations in protein structure databases like the PDB. Retrieved mutant structures can be easily analysed and compared to the wild-type through automatically superposed structures. Local mutation sites can be filtered by RMSD to identify structurally deviations upon mutation.
The method uses the local 3D micro-environment of single residues in a query protein structure to search for similar local environments in a protein structure database where the central query residue is mutated. MicroMiner returns structures with similar environments that are identical in sequence -except for the position of the mutation- and have a similar structural arrangement. These mutant structures are automatically superposed to the query wild-type protein for easy comparison and analysis of the structural changes of mutations. The method originates from the ASCONA2/SIENA3 technology.
1. Sieg, J.; Rarey, M., Searching similar local 3D micro-environments in protein structure databases with MicroMiner. Brief. Bioinform. 2023, 24 (6), bbad357. DOI: https://doi.org/10.1093/bib/bbad357
2. Bietz, S.; Rarey, M., ASCONA: Rapid Detection and Alignment of Protein Binding Site Conformations. J Chem Inf Model 2015, 55 (8), 1747-56. DOI: https://doi.org/10.1021/acs.jcim.5b00210
3. Bietz, S.; Rarey, M., SIENA: Efficient Compilation of Selective Protein Binding Site Ensembles. J Chem Inf Model 2016, 56 (1), 248-59. DOI: https://doi.org/10.1021/acs.jcim.5b00588
Run a Query
- Search for all residue positions of the protein at once by just clicking "Calculate".
- Select a list of interesting residues. This will usually run faster for larger protein complexes with >1000 residues.
LifeSoaks
LifeSoaks calculates solvent channels and their corresponding
bottleneck radii by constructing a three dimensional Voronoi
diagram.
You can use it to compute a bottleneck radius for a complete
crystal structure as well as a ccp4 grid file storing
local bottleneck radii for analyzing the structure with
respect to specific regions of interest.
When changing the cut-off radius, the displayed
grid changes as well. For the current cut-off, the visualized
grid represents the space in which a sphere with that radius
could roll around freely on an infinite path through the crystal.
When the protein strongly reaches outside its unit cell, it might
be necessary to visualize several asymmetric unit copies, which is
currently not supported in this viewer. We recommend downloading
the ccp4 file in these cases and loading it with more
sophisticated visualization programs.
1. Pletzer-Zelgert, J.; Ehrt, C.; Fender, I.; Griewel, A.; Flachsenberg, F.; Klebe, G.; Rarey, M.,
LifeSoaks: a tool for analyzing solvent channels in protein crystals and obstacles for soaking experiments.
Acta Cryst. 2023, D79, 837-856. DOI:
https://doi.org/10.1107/S205979832300582X
Settings
JAMDA
JAMDA docks small molecules into protein binding sites.
1. Flachsenberg, F.; Ehrt, C.; Gutermuth, T.; Rarey, M., Redocking the PDB. J Chem Inf Model 2023. DOI: https://doi.org/10.1021/acs.jcim.3c01573
2. Schellhammer, I.; Rarey, M., TrixX: structure-based molecule indexing for large-scale virtual screening in sublinear time. J Comput Aided Mol Des 2007, 21 (5), 223-38. DOI: https://doi.org/10.1007/s10822-007-9103-5
3. Henzler, A. M.; Urbaczek, S.; Hilbig, M.; Rarey, M., An integrated approach to knowledge-driven structure-based virtual screening. J Comput Aided Mol Des 2014, 28 (9), 927-39. DOI: https://doi.org/10.1007/s10822-014-9769-4
4. Flachsenberg, F.; Meyder, A.; Sommer, K.; Penner, P.; Rarey, M., A Consistent Scheme for Gradient-Based Optimization of Protein-Ligand Poses. J Chem Inf Model 2020, 60 (12), 6502-6522. DOI: https://doi.org/10.1021/acs.jcim.0c01095
5. Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M., Analyzing the topology of active sites: on the prediction of pockets and subpockets. J Chem Inf Model 2010, 50 (11), 2041-52. DOI: https://doi.org/10.1021/ci100241y
Settings
WarPP
WarPP is a fully automated procedure to place water molecules in the active site of a protein structure.
In the corresponding publication we showed that WarPP places 80% of water molecules within 1.0 Å distance to a crystallographically observed one.
1. Nittinger, E.; Flachsenberg, F.; Bietz, S.; Lange, G.; Klein, R.; Rarey, M., Placement of Water Molecules in Protein Structures: From Large-Scale Evaluations to Single-Case Examples. J Chem Inf Model 2018, 58 (8), 1625-1637. DOI: https://doi.org/10.1021/acs.jcim.8b00271
GeoMine
GeoMine enables textual, numerical and 3D searching with full chemical awareness
in binding sites of the entire PDB dataset.
1,2
We are aware of a problem regarding picking from the structure:
This problem does seem to only occur in Chromium-based browsers (e.g., Chrome/Edge).
Safari and Firefox are not affected.
We are working on a fix - thanks for your patience.
1. Graef, J.; Ehrt, C.; Diedrich, K.; Poppinga, M.; Ritter, N.; Rarey, M., Searching Geometric Patterns in Protein Binding Sites and Their Application to Data Mining in Protein Kinase Structures. J Med Chem 2022, 65 (2), 1384-1395. DOI: https://doi.org/10.1021/acs.jmedchem.1c01046
2. Diedrich, K.; Graef, J.; Schoning-Stierand, K.; Rarey, M., GeoMine: interactive pattern mining of protein-ligand interfaces in the Protein Data Bank. Bioinformatics 2021, 37 (3), 424-425. DOI: https://doi.org/10.1093/bioinformatics/btaa693
3. Inhester, T.; Bietz, S.; Hilbig, M.; Schmidt, R.; Rarey, M., Index-Based Searching of Interaction Patterns in Large Collections of Protein-Ligand Interfaces. J Chem Inf Model 2017, 57 (2), 148-158. DOI: https://doi.org/10.1021/acs.jcim.6b00561
StructureProfiler
StructureProfiler was developed as an all-in-one tool to screen structures based on selection criteria typically used upon dataset assembly for structure-based design methods.1
1. Meyder, A.; Kampen, S.; Sieg, J.; Fahrrolfes, R.; Friedrich, N. O.; Flachsenberg, F.; Rarey, M., StructureProfiler: an all-in-one tool for 3D protein structure profiling. Bioinformatics 2019, 35 (5), 874-876. DOI: https://doi.org/10.1093/bioinformatics/bty692 2. Hartshorn, M. J.; Verdonk, M. L.; Chessari, G.; Brewerton, S. C.; Mooij, W. T.; Mortenson, P. N.; Murray, C. W., Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem 2007, 50 (4), 726-41. DOI: https://doi.org/10.1021/jm061277y 3. Warren, G. L.; Do, T. D.; Kelley, B. P.; Nicholls, A.; Warren, S. D., Essential considerations for using protein-ligand structures in drug discovery. Drug Discov Today 2012, 17 (23-24), 1270-81. DOI: https://doi.org/10.1016/j.drudis.2012.06.011 4. Friedrich, N. O.; Meyder, A.; de Bruyn Kops, C.; Sommer, K.; Flachsenberg, F.; Rarey, M.; Kirchmair, J., High-Quality Dataset of Protein-Bound Ligand Conformations and Its Application to Benchmarking Conformer Ensemble Generators. J Chem Inf Model 2017, 57 (3), 529-539. DOI: https://doi.org/10.1021/acs.jcim.6b00613 5. Meyder, A.; Nittinger, E.; Lange, G.; Klein, R.; Rarey, M., Estimating Electron Density Support for Individual Atoms and Molecular Fragments in X-ray Structures. J Chem Inf Model 2017, 57 (10), 2437-2447. DOI: https://doi.org/10.1021/acs.jcim.7b00391
Settings
If you find the ProteinsPlus server useful, please participate in our survey and help us to further improve our services.